ვილსონის დაავადება არის გენეტიკური დარღვევა, რა დროსაც ორგანიზმში ირღვევა სპილენძის დონე და განსაკუთრებით დიდი რაოდენობით აკუმულირდება თავის ტვინსა და ღვიძლში.მკურნალობის გარეშე დაავადებამ შესაძლოა გამოიწვიოს სიცოცხლისთვის საშიში პრობლემები.მკურნალობა მედიკამენტოზურია და გულისხმობს უკვე ჩალაგებული სპილენძის მოცილებას ან უზრუნველყოფს მის პრევენციას.არის იშვიათი დაავადება და გადაეცემა მემკვიდრეობით, ვლინდება ყოველი 30 000 ადამიანიდან 1-ში.
ვილსონის დაავადების გენის მქონე ადამიანის ორგანიზმში ირღვევა სპილენძის ცვლის პროცესი.ნორმაში, ჯანმრთელობის შენარჩუნებისთვის საჭიროა სპილენძის სულ მცირე რაოდენობა, რომლის ათვისებაც ხდება სხვადასხვა საკვებიდან, დაავადების არსებობისას კი ორგანიზმი ვერ უზრუნველყოფს მისი ზედმეტი რაოდენობისგან გათავისუფლებას და ლაგდება ღვიძლში, თავის ტვინში, თირკმელებსა და რქოვანაში.დიდი რაოდენობით სპილენძის აკუმულირება ღვიძლის უჯრედებში, სახელად ჰეპატოციტებში, საზიანოა და იწვევს ღვიძლის დაზიანებას.რაც შეეხება თავის ტვინს, სპილენძი ძირითადად გროვდება ოსპისებრ ბირთვში.ვილსონის დაავადებას ასევე უწოდებენ ჰეპატოლენტიკულარულ დაგენერაციას.
დააავდებას საფუძვლად უდევს მე-13 წყვილი ქრომოსომის კონკრეტული გენის უფუნქციობა, სახელწოდებით ATP7B.სწორედ ეს გენი არის პასუხისმგებელი ღვიძლის უჯრედებიდან ზედმეტი რაოდენობის სპილენძის მოცილებაზე.ჩვეულებრივ, ღვიძლი მას ნაღვლის წვენის საშუალებით გამოყოფს ორგანიზმიდან.მაგრამ მას შემდეგ, რაც ჰეპატოციტები ამოწურავს რესურსს დაიგროვოს სპილენძი, ის გადადის სისხლის მიმოქცევის სისტემაში და ღვიძლის გარდა აკუმულირდება სხვა ორგანოებში, უპრატესად თავის ტვინში.
დაავადებას ახასიათებს ავტოსომურ-რეცესიული დამემკვიდრება, რაც ნიშანავს იმას, რომ დააავდების განვითარებისთვის საჭიროა ორი ანომალური ATP7B გენი, რომელთაგან ერთი გადაეცემა დედის, ხოლო მეორე მამისგან.ერთი ანომალური გენის არსებობის შემთხვევაში პირი არის მხოლოდ მატარებელი და არა დაავადებული, ვინაიდან მეორე ნორმალური გენი სრულიად საკმარისია იმისთვის, რომ გაკონტროლდეს სპილენძის ცვლა ორგანიზმში, თუმცა მატარებელს შეუძლია ანომალური გენი გადასცეს შთამომავლობას.
რამდენად ხშირია ვილსონის დაავადება?
100-დან 1 ადამიანი არის ანომალური გენის მატარებელი.
ორი ანომალური გენის მქონე მშობელს შესაძლოა ჰყავდეს:
4-დან 1 შემთხვევაში უილსონის დაავდების მქონე შვილი
4-დან 2 შემთხვევაში ერთი ანომალური გენის მატარებელი შვილი
4-დან 1 შემთხვევაში დაავადების და გენის არ მქონე შვილი
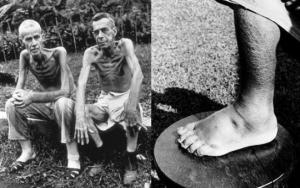
მიუხედავად იმისა, რომ გენეტიკური დეფექტი ვლინდება დაბადებისთანავე, სპილენძის დამაზიანებელი რაოდენობით აკუმულირებისთვის წლებია საჭირო, სიმპტომები ჩვეულებრივ ვლინდება 6 დან 20 წლის ასაკამდე, უფრო ხშირად თინეიჯერობისას, თუმცა შესაძლოა განვითარდეს შუახნის ასაკშიც.
დაავადების გამოვლინება უპირატესად იწყება ღვიძლისმიერი სიმპტომებით:
სიყვითლე
გულისრევა და ღებინება
აბდომინალური ტკივილი
სისუსტისა და დაღლილობის შეგრძნება
წონის კლება
მადის დაქვეითება
წყალმანკი
კუნთების ტკივილი
თავის ტვინში სპილენძის დაგროვება იწვევს როგორც ფიზიკურ, ისე მენტალურ პრობლემებს.
ფიზიკურ სიმპტომებს მიეკუთვნება:
ტრემორი
მოძრაობის შენელება
წერისა და მეტყველების სირთულეები
თავის ტკივილი
გულყრა
ყლაპვის გართულება
არამდგრადი სიარულის მანერა
მენტალური პრობლემები:
დეპრესია
ხასიათის ცვლილებები
კონცენტრაციის სირთულე
მკურნალობის უგულებელყოფამ შესაძლოა დაამძიმოს მდგომარეობა და განვითარდეს:
კუნთების ძლიერი სისუსტე
რიგიდობა
დემენცია
ვილსონის დაავადებისთვის პათოგნომური ნიშანი არის კაიზერ-ფლეიჩერის ბეჭედი - მოყავისფრო პიგმენტის არსებობა რქოვანაზე. კაიზერ-ფლეიჩერის ბეჭედი ვლინდება ვილსონის დაავდების მქონეთა 97%-ში.
სხვა კლინიკურ ნიშნებს მიეკუთვნება:
ანემია
თირკმელების დაზიანება
კარდიალური პრობლემები
პანკრეატიტი
მენსტრუაციული ციკლის დარღვევები
განმეორებითი მუცლის მოშლა
ოსტეოპოროზი
როგორ ხდება დიაგნოსტირება?
დაავადებაზე ეჭვის შემთხვევაში, დიაგნოსტირება ხდება სხვადასხვა მეთოდით:
სისხლის ანალიზი-განისაზღვრება ცერულოპლაზმინის დონე.ეს არის ცილა, რომელსაც სპილენძი უკავშირდება სისხლის მიმოქცევის სისტემაში, ვილსონის დაავადების მქონე პირებში მისი დონე დაბალია
შარდის ანალიზი-განისაზღვრება შარდში სპილენძის რაოდენობა 24 საათის განმავლობაში, ნორმაზე მაღალი მაჩვენებელი მიანიშნებს დაავადების არსებობაზე
ოფთალმოლოგიური გამოკვლევა - მნიშვნელოვანია კაიზერ-ფლეიჩერის ბეჭდის განვითარების შემოწმებისათვის
ღვიძლის ბიოფსია
თავის ტვინისა და თირკმელების MRI
ვილსონის დაავადების მკურნალობა სასიცოცხლოდ აუცილებელია.ადრეული მკურნალობა იძლევა უფრო დიდ შანსს თავიდან იქნეს აცილებული თავის ტვინისა და ღვიძლის მნიშვნელოვანი დაზიანება.
პენიცილამინი- არის ქელატური აგენტი, რომელიც უკავშირდება სპილენძს და გადააქვს ის ორგანოებიდან სისხლის მიმოქცევის სისტემაში, შემდეგ იფილტრება თირკმელებში და ზედმეტი რაოდენობის სპილენძს გამოდევნის ორგანიზიდან შარდის საშუალებით
ტრიენტინი-ასევე ქელატური აგენტია და წარმოადგენს პენიცილამინის ალტერნატივას
თუთია-არ არის ქელატური აგენტი, ის ხელს უშლის ნაწლავებში საკვებიდან სპილენძის აბსორბციას, შესაბამისად ის არ განაპირობებს ზედმეტი რაოდენობის სპილენძის გამოდევნას ორგანიზმიდან, არამედ უზრუნველყოფს სპილენძის აკუმულირების პრევენციას, ამასთან ქელატურ აგენტებთან შედარებით აქვს ნაკლები გვერდითი მოვლენა.თუთია შესაძლებელია გამოყენებულ იქნეს ადრეულ სტადიებზე, როდესაც სიმპტომატიკა ჯერ კიდევ არ ვლინდება, ასევე შეიძლება დაინიშნოს იმ პირებშიც, ვინც უკვე ჩაიტარა ქელატური აგენტებით მკურნალობა და საჭიროა სპილენძის ნორმალური დონის შენარჩუნება, ასევე შესაძლებელია დაინიშნოს ორსულებშიც.
იმ შემთხვევაში, თუ მკურნალობა უშედეგოა ან დიაგნოზი დაისვა გვიან სტადიებზე, მაშინ როდესაც კლინიკურად ვლინდება ციროზი ან ღვიძლის უკმარისობა, შესაძლებელია ჩატარდეს ღვიძლის ტრანსპლანტაცია.
მნიშვნელოვანია ყურადღება მიექცეს კვებას და რაციონიდან ამოღებულ იქნას ან მაქსიმალურად შეიზღუდოს სპილენძით მდიდარი საკვები, ასეთებია: ღვიძლი, თხილეული, სოკო და მოლუსკები.
ადრეულ სტადიებზე მკურნალობის ჩატარების შემთხვევაში პროგნოზი დადებითია, ზოგიერთი სიმპტომი სრულად ქრება, ზოგიერთი მკურნალობის შემთხვევაშიც კი შეიძლება მუდმივად დარჩეს, მაგალითად თავის ტვინის დაზიანების შემთხვევაში სიმპტომების ნაწილი ერთხელ განვითარების შემთხვევაში პერმანენტულია.მკურნალობის უგულებელყოფა კი ფატალურია.